

3D-QSAR, Molecular Docking and Molecular Dynamics Simulations of 3-Phenylsulfonylaminopyridine Derivatives as Novel PI3Ka Inhibitors
WANG Xiang-Cong, YANG Mao-Cheng, ZHANG Mo-Xuan, HU Yin-Jie, WANG Zhong-Hua* and WU Fan-Hong*
Chin. J. Struct. Chem. 2021, 40, 1567-1585 DOI: 10.14102/j.cnki.0254-5861.2011-3216
December 15, 2021
PI3K inhibitor, 3D-QSAR, molecular docking, molecular dynamics simulation
ABSTRACT
The
p110a, catalytic subunit of PI3Ka, was the primary phosphoinositide 3-kinases (PI3Ks) isoform involved in
oncogenic RTK signaling and tumorigenesis. In this study, the three-dimensional
quantitative structure-activity relationship (3D-QSAR), molecular docking and molecular dynamics simulation were
employed to study the binding mode between 3-phenylsulfonylaminopyridine derivatives
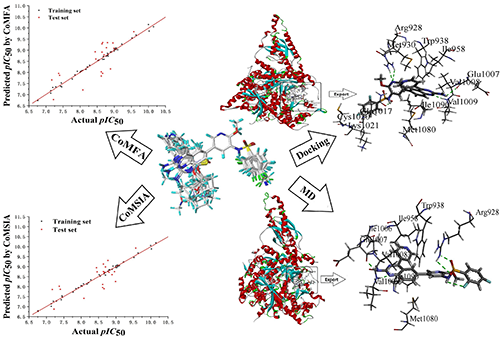
and PI3Ka. The stable and reliable 3D-QSAR models were constructed based on the
application of the comparative molecular field analysis (CoMFA) model (q2 = 0.704, r2 = 0.994) and comparative
molecular similarity index analysis (CoMSIA) model (q2 = 0.804, r2 = 0.996). The contour maps illustrated relationship between structure and
biological activity. The conformation obtained after MD simulation was more
stable than the docked conformation. MD simulation was performed in a more
realistic environment, and was much closer to physiological conditions. As a result, five novel PI3Ka inhibitors were designed
with better biological activity than the template compound 8.





- Address: Fuzhou High & New Technology Industrial Zone, Shangjie, Minhou, Fuzhou, Fujian 350108, China
- Tel: (86)-591-63173769, 63173770
- E-mail: cjsc@fjirsm.ac.cn
CN 35–1112/TQ, ISSN 0254–5861, Online ISSN: 2949-768X Copyright @ 2022 Chinese Journal of Structural Chemistry-www.Chinese Journal of Structural Chemistry.net. All Rights Reserved 闽ICP备2022002645号-1