

Cover Picture
Metal-ion-tuned metal-organic frameworks for C2H2/CO2 separation
Meng Sun, Hongyan Liu, Xiaokang Wang, Xinlei Yang, Fei Gao, Deyu Xie, Weidong Fan*, Yinfeng Han*, Ben Xu, Daofeng Sun Submit a Manuscript
Metal-ion-tuned metal-organic frameworks for C2H2/CO2 separation
Meng Sun, Hongyan Liu, Xiaokang Wang, Xinlei Yang, Fei Gao, Deyu Xie, Weidong Fan*, Yinfeng Han*, Ben Xu, Daofeng Sun Submit a Manuscript
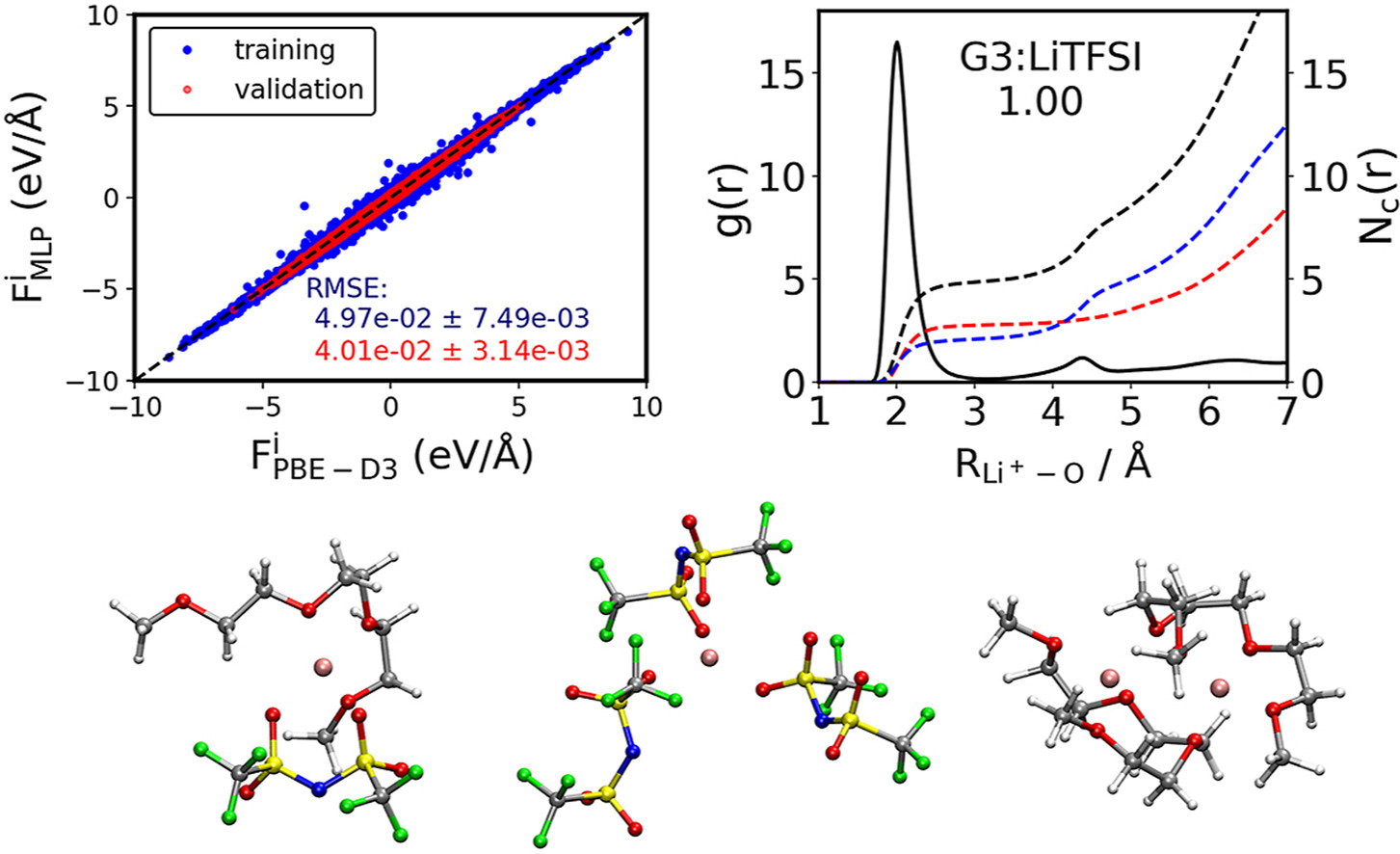
Understanding the solvation structures of glyme-based electrolytes by machine learning molecular dynamics
Feng Wang, Jun Cheng*
Chin. J. Struct. Chem., 2023, 42: 100061. DOI: 10.1016/j.cjsc.2023.100061
September 15, 2023
Glyme-based electrolytes; Machine learning potential; Molecular dynamics; Solvation structure
ABSTRACT
Glyme-based electrolytes are of great interest for rechargeable lithium metal batteries due to their high stability, low vapor pressure, and non-flammability. Understanding the solvation structures of these electrolytes at the atomic level will facilitate the design of new electrolytes with novel properties. Recently, classical molecular dynamics (CMD) and ab initio molecular dynamics (AIMD) have been applied to investigate electrolytes with complex solvation structures. On one hand, CMD may not provide reliable results as it requires complex parameterization to ensure the accuracy of the classical force field. On the other hand, the time scale of AIMD is limited by the high cost of ab initio calculations, which causes that solvation structures from AIMD simulations depend on the initial configurations. In order to solve the dilemma, machine learning method is applied to accelerate AIMD, and its time scale can be extended dramatically. In this work, we present a computational study on the solvation structures of triglyme (G3) based electrolytes by using machine learning molecular dynamics (MLMD). Firstly, we investigate the effects of density functionals on the accuracy of machine learning potential (MLP), and find that PBE-D3 shows better accuracy compared to BLYP-D3. Then, the densities of electrolytes with different concentration of LiTFSI are computed with MLMD, which shows good agreement with experiments. By analyzing the solvation structures of 1 ns MLMD trajectories, we found that Li+ prefers to coordinate with a G3 and a TFSI− in equimolar electrolytes. Our work demonstrates the significance of long-time scale MLMD simulations for clarifying the chemistry of non-ideal electrolytes.





- Address: Fuzhou High & New Technology Industrial Zone, Shangjie, Minhou, Fuzhou, Fujian 350108, China
- Tel: (86)-591-63173769, 63173770
- E-mail: cjsc@fjirsm.ac.cn
CN 35–1112/TQ, ISSN 0254–5861, Online ISSN: 2949-768X Copyright @ 2022 Chinese Journal of Structural Chemistry-www.Chinese Journal of Structural Chemistry.net. All Rights Reserved 闽ICP备2022002645号-1