

Nitrogen doping retrofits the coordination environment of copper single-atom catalysts for deep CO2 reduction
Yuxiang Zhang, Jia Zhao, Sen Lin*
Submit a Manuscript
Yuxiang Zhang, Jia Zhao, Sen Lin*
Chin. J. Struct. Chem., 2024, 43: 100415. DOI: 10.1016/j.cjsc.2024.100415
November 15, 2024
Electrocatalytic CO2 reduction; Cu single-atom catalysts; Density functional theory; Coordination environment
ABSTRACT
The electrocatalytic CO2 reduction reaction (CO2RR) represents an effective way to address energy crises and
environmental issues by converting CO2 into valuable chemicals. Single-atom catalysts (SACs) can achieve
excellent catalytic activity in CO2RR. However, the study of CO2RR on SACs still poses significant challenges,
especially in terms of controlling the selectivity towards the deep product such as CH4 and CH3OH. Herein, we
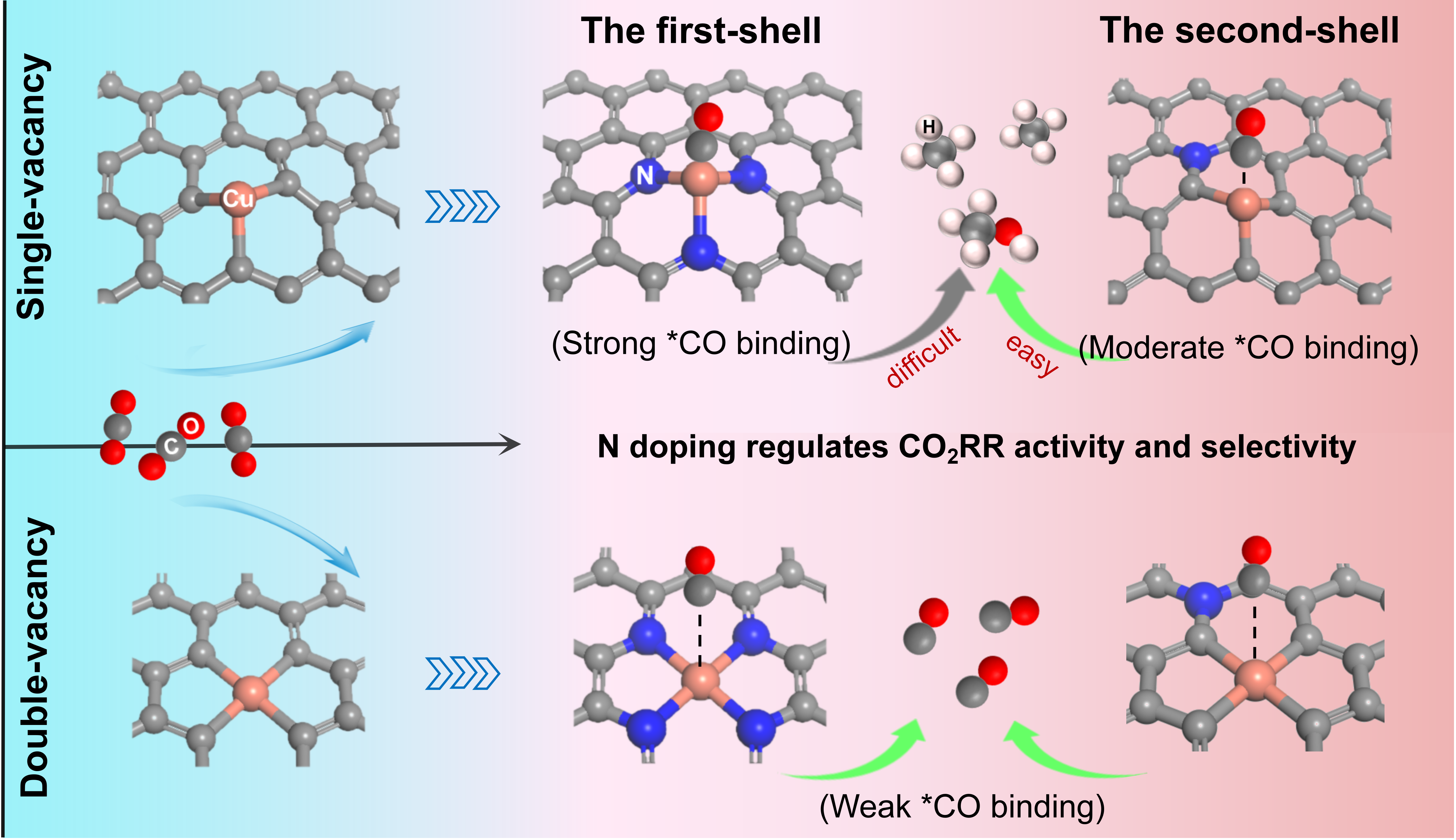
employ density functional theory (DFT) calculations to investigate the CO2RR on Cu SAC supported on N-doped
graphene (Cu-N/C) and explore the role of N dopants on the CO2RR performance. Compared to Cu SACs sup
ported on N-doped defective graphene with double vacancy (Cu-N/C-DV), Cu SACs supported on N-doped
defective graphene with single vacancy (Cu-N/C-SV) can effectively convert CO2 into the deeply reduced C1 products, including CH4 and CH3OH, thus further indicating that Cu-N/C-SV has a stronger interaction with *CO,
which is conducive to the deep reduction of *CO. Increasing the coordination number of N atoms or the proximity
of doping site to the Cu active site can effectively enhance the stability of catalyst and promote the adsorption of
*CO on Cu-N/C-SV. However, this also increases the free energy of the formation of *CHO intermediate. The
results suggest that CuC3-Nm, which contains a N atom in the second coordination shell (meta-position) of Cu
SACs, has the best electrocatalytic performance of CO2RR in terms of both selectivity and catalytic activity, not
only contributing to an in-depth understanding of the reaction mechanism of CO2RR on SACs but also providing
insights into the design of SACs for efficient CO2RR.





- Address: Fuzhou High & New Technology Industrial Zone, Shangjie, Minhou, Fuzhou, Fujian 350108, China
- Tel: (86)-591-63173769, 63173770
- E-mail: cjsc@fjirsm.ac.cn